Neurofibromatose Typ 2 (NF2)
Neurofibromatose Typ 2 (NF2)
Was ist Neurofibromatose Typ 2?
Die Neurofibromatose Typ 2 (NF2) ist eine Erkrankung, die durch die Entwicklung gutartiger Tumore entlang der Nerven gekennzeichnet ist, die als Schwannome bezeichnet werden und fast immer an den Hör- und Gleichgewichtsnerven auftreten (vestibuläre Schwannome-VS). Die meisten NF2-Patienten entwickeln VS auf beiden Seiten, was zu Gleichgewichtsproblemen und gegebenenfalls zu völliger Taubheit führen kann. Schwannome an anderen Nerven im Kopf, in der Wirbelsäule und weiteren Bereichen des Körpers können zu einem Verlust der Muskelfunktion und der Sensitivität führen, wodurch viele NF2-betroffene Menschen im Verlauf auf einen Rollstuhl angewiesen sind. Der zweithäufigste Tumor bei NF2 ist ein gutartiger Tumor der Gehirnhäute und des Rückenmarks, der als Meningeom bezeichnet wird. Auch dies kann zu einem Verlust der normalen Funktion führen sowie zu Kopfschmerzen und Krampfanfällen. Zudem betrifft ein Tumor im Rückenmark, der als Ependymom bezeichnet wird, etwa 20-40 % der Menschen, ist aber in der Regel nicht fortschreitend. NF2 ist eine Erkrankung die zu Einschränkungen und Limitierung des Lebens führen kann.
Was verursacht Neurofibromatose Typ 2?
NF2 ist eine erblich bedingte genetische Erkrankung, die durch eine Veränderung auf einer Kopie des NF2-Gens auf Chromosom 22 verursacht wird.
Wie wird Neurofibromatose Typ 2 vererbt?
NF2 wird in etwa 40 % der Fälle von einem betroffenen Elternteil vererbt. In den restlichen Fällen entsteht die NF2-Genveränderung bei dieser Person neu. Diese NF2-Veränderung ist entweder in allen Zellen vorhanden (65-70 %), was darauf hinweist, dass die Veränderung in der Eizelle oder dem Spermium vorhanden war, als diese bei der bei der Empfängnis verschmolzen sind, oder nur in einigen Zellen, was darauf hinweist, dass die genetische Veränderung während der Entwicklung des Embryos aufgetreten ist (sog. Mosaik). Die Mosaik-NF2 ist milder, da nicht alle Zellen betroffen sind und die Wahrscheinlichkeit, NF2 an Kinder weiterzugeben ist geringer als die üblichen 50 %. Die Identifizierung des zugrunde liegenden Gendefekts ist auch hilfreich, um eine Prognoseabschätzung abgeben zu können. In Einzelfällen wird mit der genetischen Analyse des Tumorgewebes begonnen, anstatt mit der genetischen Analyse aus Blut.
Welche Vorsorgemöglichkeiten gibt es?
Das Screening auf NF2 kann mit dem Testen noch nicht betroffener Verwandter beginnen, bevor Symptome für die in der Familie vorliegende genetische Veränderung auftreten. Danach basiert das Screening im Allgemeinen auf der klinischen Symptomatik, falls vorhanden, sowie eine jährliche MRT-Schädel-Untersuchung und alle drei Jahre eine MRT-Untersuchung der Wirbelsäule. Die Therapie umfasst die chirurgische Entfernung von Tumoren, eine medikamentöse Behandlungen mit Bevacizumab bei schnell wachsenden Schwannomen und in ausgewählten Fällen die gezielte Strahlenbehandlungen (nicht im Kindesalter).
Leitlinie für die klinische Praxis
Unterstützt von ERN GENTURIS*
* ERN GENTURIS nutzt AGREE II als Instrument zur Bestätigung von Leitlinien. Die Qualität der Leitlinie wird durch die Bewertung der Gründlichkeit und Transparenz des Leitlinienentwicklungsprozesses bewertet. Der Inhalt der Leitlinie wird nicht bewertet, obwohl die Auswahl der Leitlinie für die Anerkennung eine Expertenmeinung für den Nutzen des Inhalts der Leitlinie beinhaltet.
Klinische Versorgung
Versorgungspfad - Neurofibromatose Typ 2
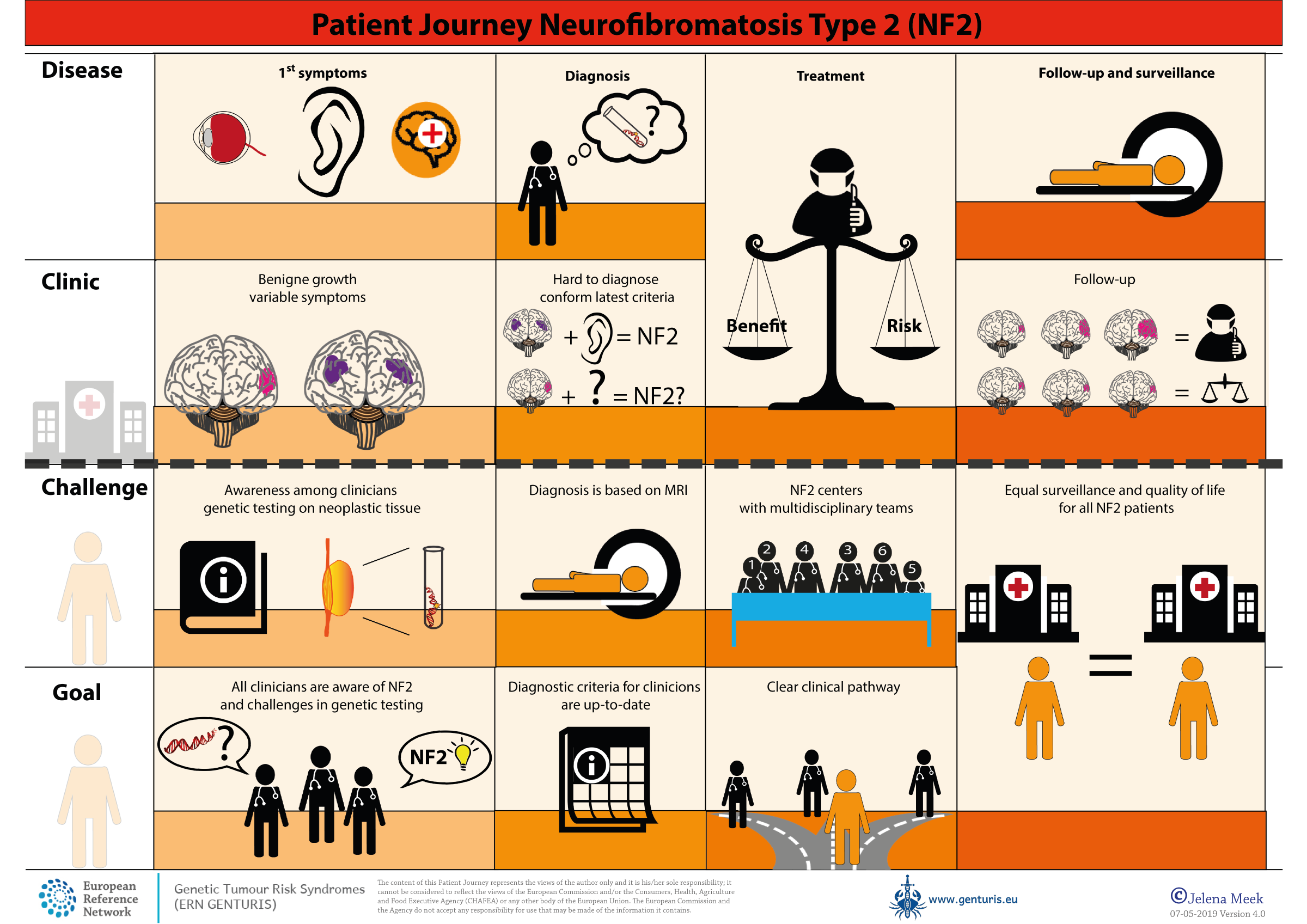
Patientenreise
Patientenreise - Neurofibromatose Typ 2